全国服务热线400-689-6719
细胞平台
RNA甲基化位点分析新方法
继上次QB期刊给大家推荐了WendyV. Gilbert教授的《关于pre-mRNA存在的修饰形式及其对剪接的影响》综述后,小编看到读者们的热情度特别高。借此机会,再给大家推荐一篇来自德克萨斯大学西南医学中心,QBRC、BICF中心主任谢阳教授实验室在QB期刊上发表的最新关于分析MeRIP-Seq (methylated RNA immunoprecipitation sequencing)数据的新方法(A Bayesian hierarchical model for analyzing methylated RNA immunoprecipitation sequencing data)。(RNA甲基化)
在《A Bayesian hierarchical model for analyzing methylated RNA immunoprecipitation sequencing data》这篇文章中,我们首次提出用一种贝叶斯统计模型,即贝叶斯层次模型BaySeqPeak,用于分析MeRIP-Seq数据,从而帮助研究人员发现转录组中的甲基化位点信号[1]。

RNA甲基化数据分析现状
DNA与组蛋白的表观遗传修饰在调控基因表达上的重要影响已为科学界所广泛熟知。同DNA一样,作为生物遗传信息传递中的重要一环,RNA分子也广泛存在着化学修饰。目前,科学家已经鉴定确认了超过100种的RNA化学修饰方式,其中以m6A(N6-methyladenosine,6-甲基腺嘌呤,化学结构见图1)最为常见[2]。
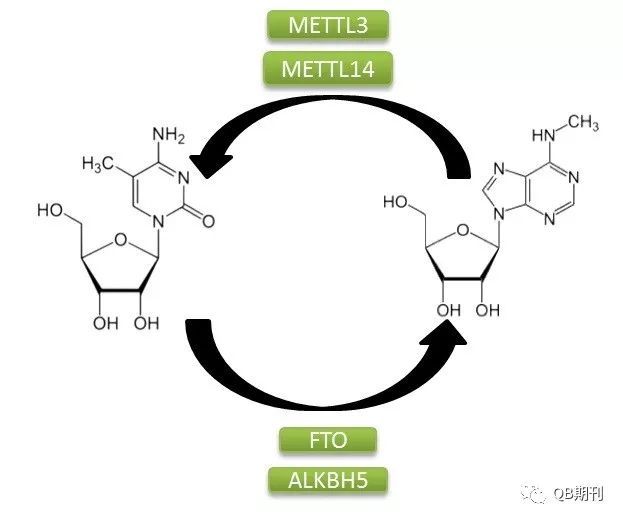
图1 m6A甲基化修饰过程
m6A甲基化修饰是一种由多种蛋白参与的动态可逆的修饰方式。它的生成主要是由甲基转移酶复合体介导,其中包含METTL3,METTL14和WTAP;而擦除甲基化修饰基团的过程则由去甲基化酶FTO和ALKBH5负责。此外,多种蛋白,如YTHDF1和YTHDF3都可识别m6A信号位点,并通过结合下游效应蛋白的方式传递甲基化信号。目前已经发现,m6A在调控基因表达、剪接、RNA 编辑、RNA 稳定性和控制mRNA寿命和降解等多方面都存在重要的影响[3]。
虽然RNA甲基化在上世纪七十年代就已经被发现证实,但长期以来由于技术局限,相关的修饰机理、调控手段以及生物学意义一直未能阐明。现在,MeRIP-Seq (methylated RNA immunoprecipitation sequencing)技术的出现[4,5](图2),使得通过高通量手段在全转录组(transcriptome)水平上研究m6A甲基化修饰变为可能。
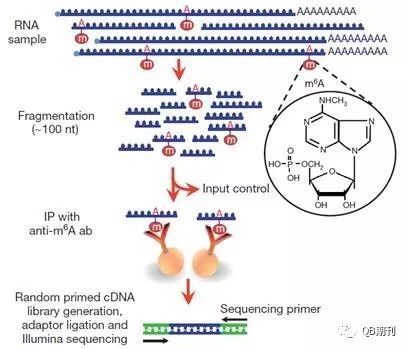
图2 MeRIP-Seq技术流程图
通行的分析MeRIP-Seq数据的思路是,利用一个特定长度的(通常为100~200nt长)窗口从前至后扫描整条染色体,并记录每个样本落入每个窗口中的RNA短序列数目(read count)。通常,实验条件下(IP)样本的RNA短序列应大致分布在甲基化位点附近,而对照条件下(INPUT)则和正常的每个基因的表达值正相关(没有甲基化影响)。这一数据特点使得传统分析DNA甲基化的工具无法很好地胜任RNA甲基化数据的分析。总而言之,转化为统计语言就是,我们需要寻找那些在实验条件下序列数目显著高于对照条件下序列数目的窗口(甲基化位点),并相应地给予显著性统计值(p值或假阳性概率)。
BaySeqPeak模型分析RNA甲基化数据的优势
我们建立的BaySeqPeak模型则主要从MeRIP-Seq数据的重复样本数少,样本数据空间上前后相关,以及存在大量零数据的特点出发,利用以下三种不同的策略解决了这些难题:
1)采用零膨胀的负二项分布拟合单样本的序列计数,以防止大量零数据和过度离散破坏模型稳定性;
2)采用隐马尔可夫模型模拟单样本空间上的前后相关性;
3)利用贝叶斯统计的思路,使得模型在低样本数的条件下依然维持足够的准确度。
在模拟数据中,BaySeqPeak能很好地预测了实验人员预先设定的甲基化位点,而比较的exomePeak和MeTPeak模型则汇报了较多的假阳性和假阴性位点(图3)。
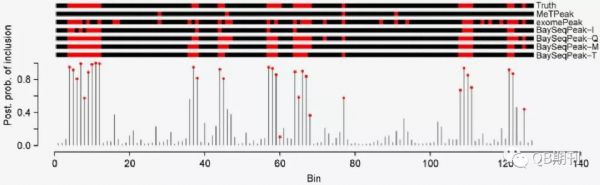
图3 模拟数据中真实的甲基化位点与各模型预测的甲基化位点(红色)
通过ROC曲线可以发现,不同参数下的模拟数据中,BaySeqPeak模型的预测准确性均显著高于exomePeak和MetPeak模型(图4)。
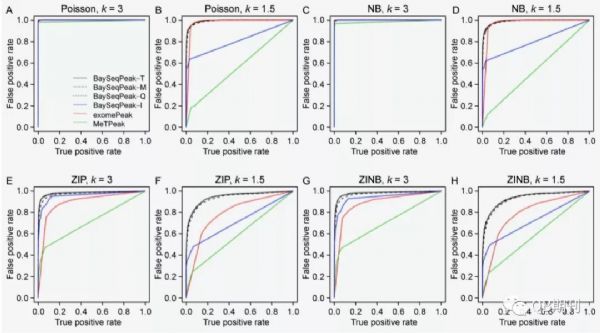
图4 不同参数条件下,各模型预测的ROC曲线
在数值收敛方面,模型在经过多次迭代之后,预测值已稳定地收敛到了真实值附近(图5)。
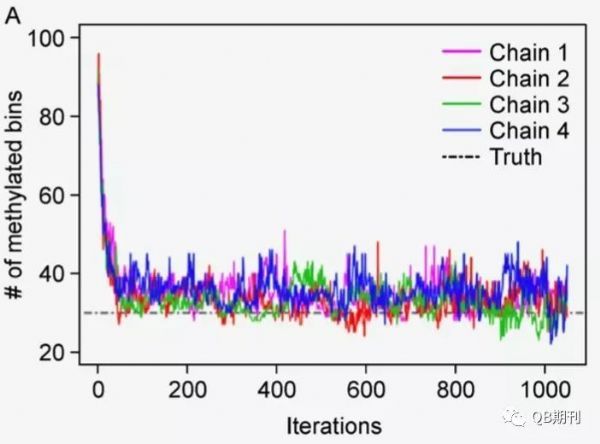
图5 甲基化位点的预测数的收敛过程
在真实数据中,BaySeqPeak模型也很好地预测了甲基化的区段。不仅如此,相较于exomePeak,BaySeqPeak还详细区分出了一个甲基化区域中临近的几个甲基化峰位,显示了模型的高精度与高分辨率。
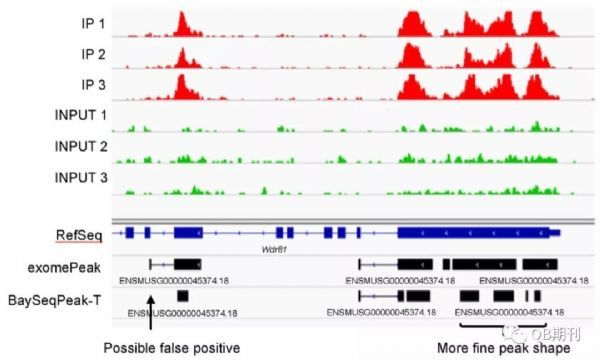
图6 一个真实数据中预测的甲基化区域
RNA甲基化的研究目前仍然处于起步阶段,修饰调控过程的具体细节,以及这些修饰如何具体地影响细胞的功能,特别是在疾病条件下,这些化学修饰是如何发生变化的仍然存在大量未知。本文提出的统计方法为有效准确地分析m6A甲基化数据提供了可能,我们期待在未来RNA甲基化的研究中能够在此模型基础上再推进一步。
参考文献
Zhang, M., Li, Q., & Xie,Y. (2018). A Bayesian hierarchical model for analyzing methylated RNA immunoprecipitationsequencing data. Quantitative Biology, 6(3), 275-286.
Machnicka, M. A., Milanowska,K., Oglou, O., Purta, E., Kurkowska, M., Olchowik, A., Januszewski, W.,Kalinowski, S., Dunin-Horkawicz, S., Rother, K. M., et al. (2013) MODOMICS: adatabase of RNA modification pathways–2013 update. Nucleic Acids Res., 41,D262–D267
Meyer, K. D., and Jaffrey, S.R. (2014) The dynamic epitranscriptome: N 6-methyladenosine and gene expressioncontrol. Nat. Rev. Mol. Cell Bio., 15, 313–326
Dominissini, D.,Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg,S., Cesarkas, K., Jacob-Hirsch, J., Amariglio, N., Kupiec, M., et al. (2012)Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature485, 201–206.
Meyer, K. D., Saletore, Y.,Zumbo, P., Elemento, O., Mason, C. E. and Jaffrey, S. R. (2012) Comprehensiveanalysis of mRNA methylation reveals enrichment in 3′ UTRs and near stopcodons. 1517 Cell, 149, 1635–1646.
(本文来源生物谷)
想要了解更多请关注“中洪博元医学实验帮 ”
上一篇:多篇文章共同解读癌细胞的“狡诈”特性!
下一篇:NEJM:临床试验首次表明联合使用免疫疗法和化疗显著延长三阴性乳腺癌患者的寿...
上海展辉生物技术有限公司
地址:上海市浦东新区东方路
联系电话:400-689-6719
联系邮箱:3369352092@qq.com
联系我们:
4006896719

扫一扫
关注东洪博元更多动态

